|
|
Lymphoide Neoplasien
Lymphom
- maligne Proliferation von Lymphozyten. Bei einem Lymphom werden vor
allem Lymphknoten betroffen. Wenn peripheres Blut oder Knochenmark
betroffen sind, dann spricht man von einer Leukämie.
Man teilt Lymphome in 2 Gruppen - immature (Vorläufezellen) und mature
(reife Zellen). Auch gibt es B-Zell-, T-Zell- und NK-Zellenlymphome.
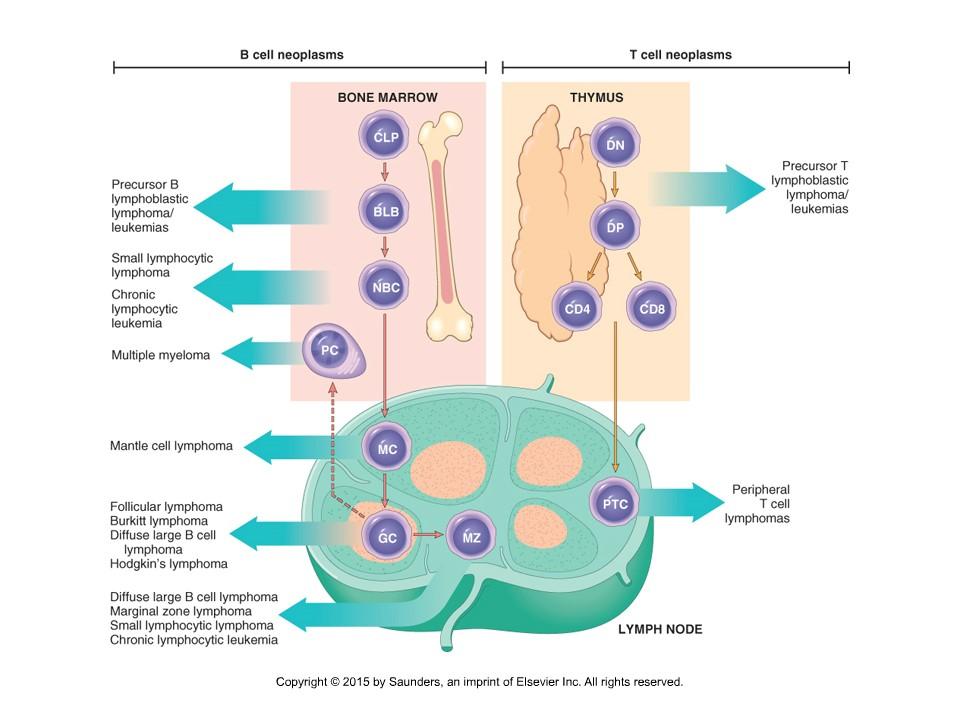
Fig. Origin of lymphoid neoplasms.
B-Zellen.
Vorläufer B-Zelle verlässt das Knochenmark und geht ins Blut. Diese
Zellen entwickeln IgM und IgD auf ihrer Oberfläche, haben CD5 und
heißen naive B-Zellen. Aus ihnen entwickelt sich mantle cell lymphoma.
Dann gehen diese Zellen ins germinativen Zentrum --> Zentroblasten,
die reifen weiter zu kleineren Zellen - Zentrozyten. Sie haben Marker -
BCL-6 und CD10. Die Zentroblasten sind origin of Diffuses großzelliges B-Lymphom (diffuse large B-cell lymphoma) und Zentrozyten - Follikuläres Lymphom (follicular lymphoma). Dann B-Zellen gehen ins Marginalzone. Hier entwickeln sich marginal zone lymphomas (splenic marginal zone lymphoma and MALT lymphomas). Wieter gehen die Zellen ins Blut. Hier entwicken sich CLL oder Plasmozytom.
Akute lymphatische Leukämie
Vorläufer B-Zelle akute lymphoblastische Leukämie (B-ALL)
Vor allem Kinder < 6 Jahre.
Immunphänotypisierung - CD10, CD19, TdT.
Klinik - Anämie, Infektionen, Blutungen. ZNS ist oft betroffen. Knochenschmerzen sind das frühere Zeichen.
Therapie - Chemotherapie. Patienten, die resisen zu Chemo sind, brauchen Knochenmarktransplantation. Die Prognose ist gut.
Vorläufer T-Zelle akute lymphoblastische Leukämie (T-ALL)
Es kommt in jedem Alter vor. Im Unterschied zu B-ALL werden mediastinale Lymphknoten betroffen (Lymphom).
Immunphänotypisierung - CD7, CD2.
Periphere (mature) B-Zellen-Lymphome
Maligne Proliferation der B-Zelle, wenn sie das Knochenmark verlassen
hat, heisst periphere Lymphome. Es gibt verschiedene B-Zellen-Lymphome,
abhängig vom Stadium der B-Zelle.
Die häufigsten sind:
- diffuse large B-cell lymphoma - 30%
- follicular lymphoma - 22%
- MALT-Lymphoma - 7%
- chronic lymphocytic leukemia - 7%
- mantle cell lymphoma - 6%.
Periphere B-Zellen-Lymphome bis auf diffuse large B-Cell lymphoma und Burkitt lymphoma sind selten bei Kindern.
Mantelzelllymphom
Eine seltene Ernkrankung. 6% aller B-Zellen-Lymphome. Marker - CD5. Positiv für cyclin D1.
Sie sind agressiv und schwer zu behandeln.
Lebenserwartung - 3-5 Jahre.
Diffuses grosszelliges B-Lymphom
Marker - CD19, CD20. Negativ für TdT, cyclin D1.
DLBCL ist agressiv, aber behandelbar.
Burkitt Lymphom
Burkitt Lymphom wächst schnell.
Ursache - chromosomal Aberration. --> Myc Gen
3 Typen:
- endemisches Burkitt Lymphom - in Africa. Gesichtsknochen sind
betroffen. Bei Kindern. Fast bei allen kann man Virus Epstein Barr
finden.
- sporadisches BL - oft bei Kindern, überall in der Welt. Oft manifestiert sich mit Raumforderung im Bauch.
- Immundefizit-assoziiertes BL - bei HIV-Infizierten.
Pathologie. Eher extranodal.
BL wächst schneel und deswegen sensibel zur Chemotherapie.
Follikuläres Lymphom
Chromosomal Aberration --> Bcl-2 Protein (Apoptosis Inhibitor).
Man kann es nicht behandeln. Prognosis - 7-9 Jahre, abhängig von der Differenzierung.
Spizifische Therapie - anti-CD20 - Rituximab.
B-Zelle chronische lymphatische Leukämie/kleinzelliges lymphatisches Lymphom
Population von kleinen Lymphozyten. Wenn Blut und Knochenmark betroffen
sind, dann handelt sich um eine CLL (chronische lympatische Leukämie).
Wenn Lymphknoten oder extranodale Organe betroffen sind, dann spricht
man von SLL (kleinzelliges lymphatisches Lymphom).
Pathologie. Lymphknoten sind mit monomorphen kleinzelligen Lymphozyten.
Im Blut - absolute Lymphozytose.
Prognosis - etwa 4-6 Jahre, abhängig von der Chromosomaberrationen.
Therapie. Die Patienten mit stabiler Leukozytenzahl brauchen keine Therapie. Die Leukämie ist nicht kurierbar. Rituximab.
Plasmazelluläre Neoplasien.
Plasmazelluläre Neoplasien
- monoklonale Proleferation reifer Plasmazellen, die monoklonale
Immunoglobine bilden (bösartige Vermehrung der Plasmazellen
(antikörper-produzierende B-Zellen).
Klassifizierung:
- MGUS - monoklonale Gammopathie unklarer Signifikanz - gutartige
begrenzte Vermehrung von Plasmazellen (< 10% Plasmazellen im
Knochenmark), keine Skelettdestruktionen. No CRAB (hypercalcemia, renal
insufficiency, anemia, bone lesions).
- multiples Myelom (Plasmozytom, Morbus Kahler) - Tumor der Plasmazellen mit Knochendestruktionen und monoklonalem Immunoglobin.
- Plasmazellentumor - Plasmazellen bilden einen Tumor, im Knochen oder außerhalb von Knochen (ossär oder extraossär).
- Plasmazellenleukämie - im Blut >20% maligne Plasmazellen.
Plasmozytom (multiples Myelom)
Pathologie.
1). Skelettdestruktion. Bei Osteolyse kommt es zur Hyperkalziämie.
2). Knochenmarkdysfuktion (Anämie).
3). Niereninsuffizienz - Imunnoglobulin bilden Zylinder in Tubuli.
4). Amyloidose - L-Ketten bilden Amyloid Protein (AL Amyloid) --> Ablagerung im Herzen, Nieren, Haut
Klinik. Symptome - CRAB - CRAB - hypercalcemia, renal insufficience, anemia, bone lesions.
Häufige Igs - IgG (55%) und IgA (20%). Selten IgM, IgD.
Hauptkriterin des Plasmozytoms -- 1) Immunoglobuline im Blut/Urin
(Elektrophorese) 2) Osteolysen 3) erhöhte Plasmazellen im Blut. 2 von 3
positiv für die Diagnose.
Bence-Jones Protein. -- Immunoglobuline bestehen aus 2 leichten und 2
schweren Ketten. Dieser Protein hat excessive leichte Ketten. Wichtig -
er ist nierendurchgängig.
Plasmozytom ist nicht heilbar.
Morbus Hodgkin
Morbus
Hodgkin. Histologie. -- einkernige Hodgkin-Zellen und vielkernige
Reed-Sternberg-Riesenzellen. Es ist schwer, sie zu finden. Nur 1% von
allen Zellen.
Ursprung - B-Zelle im Keimzentrum von Lymphknoten. Morbus Hodgkin betrifft auch junge Menschen, 20-30-jährige.
Morbus Hodgkin. Histologische Typen --
1). Nodulär-sklerosierende Form. Von der Kapsel an gehen Septen in den Lyphmknoten. Es bilden sich auch so Knoten.
2). Misch-Form
3). Lymphozytenreiche Form
4). Lymphozytenarme Form.
Morbus Hodgkin. Stadien. --
Stadium I - eine Gruppe von LK
II - einige Gruppen, aber auf der einen Seite von Zwechfell
III - beidseites des Zwerchfells
IV - disseminierter Befall.
Stadien I und II - Radiotherapie, III und IV - Chemotherapie.
Klinik. Vergrößerte Lymphknoten, vor allem zervikale und mediastinale. B-Symptome - 40% Patienten.
Morbus Hodgkin. Therapieschema. -- ABVD. Adriamycin, Bleomycin, Vinblastin, Dacarbazin.
Mems.
Chronische lymphatische Leukämien -- gehören zu den NHL (Non-Hodgkin-Lymphome).
CLL. Einteilung -- nach Binet A,B,C.
A < 3 vergrösserte LK-Regionen
B > 3 vergrösserte LK-Regionen
C - Hb < 10 g/dl, TZ < 100
CLL. Therapie. -- relativ gute Prognose, die Therapie ist zurückhaltend.
Tartrat-resistente saure Phosphatase -- bei Haarzellenleukämie.
Morbus Hodgkin. Histologie. -- einkernige Hodgkin-Zellen und vielkernige Sternberg-Riesenzellen.
Morbus Hodgkin. Histologische Typen --
1). Lymphozytenreiche Form
2). Nodulär-sklerosierende Form
3). Misch-Form
4). Lymphozytenarme Form.
Morbus Hodgkin. Stadien. --
Stadium I - eine Gruppe von LK
II - einige Gruppen, aber auf der einen Seite von Zwechfell
III - beidseites des Zwerchfells
IV - disseminierter Befall.
Stadien I und II - Radiotherapie, III und IV - Chemotherapie.
Morbus Hodgkin. Therapieschema. -- ABVD. Adriamycin, Bleomycin, Vinblastin, Dacarbazin.
Plasmozytom. Synonyme. -- Morbus Kahler, multiples Myelom. Tumor der Plasmazellen.
Hauptkriterin des Plasmazytoms -- 1) Immunoglobuline im Blut/Urin
(Elektrophorese) 2) Osteolysen 3) erhöhte Plasmazellen im Blut. 2 von 3
positiv für die Diagnose.
Plasmazellentumor (solitary plasmacytoma) - Plasmazellen bilden einen
Tumor im Knochen (ossär) oder extraossär (häufig Atemwege).
Plasmazellenleukämie - im Blut >20% maligne Plasmazellen
Multiples Myelom. Kalzium -- Osteolyse --> Hyperkalziämie
Multiples Myelom. Symptome - CRAB - CRAB - hypercalcemia, renal insufficience, anemia, bone lesions
Bence-Jones Protein. -- Immunoglobuline bestehen aus 2 leichten und 2
schweren Ketten. Dieser Protein hat excessive leichte Ketten. Wichtig -
er ist nierendurchg�ägig.
|
|
|